Project:Sex typing with amelogenin: Difference between revisions
Mycoplasma (talk | contribs) |
|||
| (27 intermediate revisions by 5 users not shown) | |||
| Line 1: | Line 1: | ||
= General = | |||
The general plan, with thanks to Maria, is: obtain dna samples, extract dna, pcr dna with amelogenin primers, and then run that on agarose gel. | The general plan, with thanks to Maria, is: obtain dna samples, extract dna, pcr dna with amelogenin primers, and then run that on agarose gel. | ||
| Line 11: | Line 11: | ||
It might help to try out some parts, such as the extraction, before hand, although I don't see how we know whether that works until we run it through the rest. We could practise making gels, running a ladder on them, to figure out mixtures and timings and practise with detecting the dye. | It might help to try out some parts, such as the extraction, before hand, although I don't see how we know whether that works until we run it through the rest. We could practise making gels, running a ladder on them, to figure out mixtures and timings and practise with detecting the dye. | ||
= Sampling = | |||
Current plan is to sterilise plastic sticks for consenting adult volunteers to rub inside their cheeks. Legally (I am so not a lawyer, and this is not legal advice), reading them a written spiel and getting their verbal agreement with witnesses and seeing them take the sample seems to be sufficient , although the suggestion has been made that a simple signed form might help avoid a possible problem with forgetful witnesses. | Current plan is to sterilise plastic sticks for consenting adult volunteers to rub inside their cheeks. Legally (I am so not a lawyer, and this is not legal advice), reading them a written spiel and getting their verbal agreement with witnesses and seeing them take the sample seems to be sufficient , although the suggestion has been made that a simple signed form might help avoid a possible problem with forgetful witnesses. | ||
| Line 17: | Line 17: | ||
cotton swabs are another possible option. | cotton swabs are another possible option. | ||
= Extraction = | |||
There are a number of written protocols out there | There are a number of written protocols out there | ||
| Line 35: | Line 35: | ||
The sampling and extraction are crucial steps if the following steps are to work. dna contamination can be amplified in the pcr stage. there is a suggestion that there is an amount of black art in getting this to work well. It might repay the effort to try several different extractions and run them side by side for comparison. | The sampling and extraction are crucial steps if the following steps are to work. dna contamination can be amplified in the pcr stage. there is a suggestion that there is an amount of black art in getting this to work well. It might repay the effort to try several different extractions and run them side by side for comparison. | ||
= PCR = | |||
| Line 69: | Line 69: | ||
We have SYBR green to replace the ethidium bromide. | We have SYBR green to replace the ethidium bromide. | ||
= Gel electrophoresis = | |||
== Procedure == | |||
Eng has | Eng has | ||
| Line 91: | Line 91: | ||
[http://www.shef.ac.uk/content/1/c6/06/14/01/CoSHH%20059_PCR-TGGE%20and%20geldoc.pdf Example COSHH for PCR and gel electrophoresis with TAE] | [http://www.shef.ac.uk/content/1/c6/06/14/01/CoSHH%20059_PCR-TGGE%20and%20geldoc.pdf Example COSHH for PCR and gel electrophoresis with TAE] | ||
== Stains == | |||
[http://www.nbsbio.co.uk/product.asp?pID=2490&c=283957 SafeView] and [http://www.nbsbio.co.uk/product.asp?pID=6241&cID=71 SafeWhite] are from [http://www.nbsbio.co.uk/ NBS Bio]. There are manuals and safety sheets there. | [http://www.nbsbio.co.uk/product.asp?pID=2490&c=283957 SafeView] and [http://www.nbsbio.co.uk/product.asp?pID=6241&cID=71 SafeWhite] are from [http://www.nbsbio.co.uk/ NBS Bio]. There are manuals and safety sheets there. | ||
| Line 97: | Line 97: | ||
interesting doc about ethidium bromide disposal in th uk here: [http://www.docs.csg.ed.ac.uk/estatesbuildings/waste/Ethidium_Bromide_briefing.pdf] | interesting doc about ethidium bromide disposal in th uk here: [http://www.docs.csg.ed.ac.uk/estatesbuildings/waste/Ethidium_Bromide_briefing.pdf] | ||
== What we are looking for == | |||
We expect bands at X = 977 bp and Y = 788 bp | We expect bands at X = 977 bp and Y = 788 bp | ||
| Line 110: | Line 110: | ||
It seems that the lines at X=977bp are in different positions in the male and female lanes in the cybertory example. discuss. | It seems that the lines at X=977bp are in different positions in the male and female lanes in the cybertory example. discuss. | ||
= Results = | |||
Individuals with deletions such that this test doesn't work on them are extremely rare. So we hope to get correct results for all samples. We discussed blinding the samples to avoid possible bias in interpretation reading the gel, but if we can photograph the gel that could serve that purpose. | Individuals with deletions such that this test doesn't work on them are extremely rare. So we hope to get correct results for all samples. We discussed blinding the samples to avoid possible bias in interpretation reading the gel, but if we can photograph the gel that could serve that purpose. | ||
| Line 116: | Line 116: | ||
It might be nice to do video recording and a write up. | It might be nice to do video recording and a write up. | ||
== First gel run, Dec 7, 2011 == | |||
On this date, we had not yet received our PCR tubes, so we were unable to do PCR. Instead, we decided to test our ability to do electrophoresis, using a DNA ladder and SafeWhite stain. We followed the following protocol: | On this date, we had not yet received our PCR tubes, so we were unable to do PCR. Instead, we decided to test our ability to do electrophoresis, using a DNA ladder and SafeWhite stain. We followed the following protocol: | ||
| Line 141: | Line 141: | ||
We resolved to make the next test a simple visualisation test: combine ladder with Safewhite and attempt to visualise under UV. | We resolved to make the next test a simple visualisation test: combine ladder with Safewhite and attempt to visualise under UV. | ||
== SafeWhite test, Dec 9, 2011 == | |||
Nicholas combined 2.5ul SafeWhite and 2.5ul DNA Marker-D. He was unable to make this mixture glow under UV. | Nicholas combined 2.5ul SafeWhite and 2.5ul DNA Marker-D. He was unable to make this mixture glow under UV. | ||
== Second gel run, Dec 13, 2011 == | |||
Nicholas performed the same protocol as in the "First gel run, Dec 7, 2011", with the following changes: | Nicholas performed the same protocol as in the "First gel run, Dec 7, 2011", with the following changes: | ||
| Line 156: | Line 156: | ||
* No staining was observed. | * No staining was observed. | ||
== 3rd or 4th gel run, Jan 25, 2012 == | |||
We successfully visualised a ladder using the following protocol: | We successfully visualised a ladder using the following protocol: | ||
| Line 176: | Line 176: | ||
* Visualised by placing a fluorescent tube with peak emission at 300 nm directly underneath the gel. | * Visualised by placing a fluorescent tube with peak emission at 300 nm directly underneath the gel. | ||
== 1st Feb 2012 == | |||
our Technical datasheet gives numbers for water to add to rehydrate our primers to 100uM. | |||
=== 8th Feb 2012 === | == 8th Feb 2012 == | ||
<gallery widths=160px heights=160px > | |||
File:Gel_IMG_1081_rotated.JPG|Nice picture | |||
File:Gel_IMG_20120208_223330_rot.jpg|not such a good image | |||
File:Gel_IMG_20120208_223330_crop_contrast_rot.jpg|after crop and contrast stretch | |||
File:Gel_IMG_1081_ladder.JPG|Everybody's looking for the ladder | |||
File:Gel_IMG_1081_ladder_stretch-contrast.JPG|after contrast stretch | |||
File:Gel_IMG_1081_ladder_desat_stretch-contrast.JPG|after desaturate and contrast stretch | |||
File:Gel_IMG_1081_ladder_compare.JPG|hmm... | |||
</gallery> | |||
== 12th Feb 2012 == | |||
<gallery widths=160px heights=160px > | |||
File:2012-02-12-cell-boiling.jpg|Cell lysis. | |||
File:2012-02-12-biohacking-equipment.jpg|Equipment for PCR and electrophoresis. | |||
File:2012-02-12-biohacking-electrophoresis-1.jpg|After 20 minutes of electrophoresis bands were observed in lanes 2, 3, 4, 6, and 7. The ladder was also visible in lane 5 but can't be seen in this photo. | |||
File:2012-02-12-biohacking-electrophoresis-2.jpg|200px|thumb|none|After 45 minutes of electrophoresis the bands had become very blurry. | |||
File:2012-02-12-biohacking-electrophoresis-2b.jpg|200px|thumb|none|After 45 minutes of electrophoresis, close-up. The ladder is faintly visible. | |||
File:2012-02-12-biohacking-electrophoresis ladder-comparison.jpg|On the left is the gel from 8th Feb 2012. The lines in red are clearly lines in the ladder, the orange lines are putative ladder (less certain), and the green lines are the bands. If the interpretation presented here is correct, then this result repeats the results of last Wednesday. | |||
</gallery> | |||
'''Discussion:''' | |||
* The bands are very blurry and wonky, as if the agar was of varying densities throughout. | |||
* Bands on lanes 3, 4, and 6 travelled less far than bands on lanes 2 and 7. Lane 2 did not include template (i.e. DNA); and I ran out of template in lane 7. So this is an encouraging result. | |||
* The bands are within the bounds of the ladder this time. | |||
* Bands on lanes 3, 4, and 6 are more blurry than bands on lanes 2 and 7. One exciting theory is that bands on those lanes actually worked, and the blurriness is because of the 200 base-pair difference between the two amelogenin sequences; whereas the bands on lanes 2 and 7 contain un-used primer and master mix, which is a) shorter sequences and b) more well-defined (presumably because there is less of a difference between the lengths). | |||
More details on the days results and our discussions: [[/results-20120212]] | |||
[https://lists.kentgeek.org/pipermail/lhs-biohacking/2012-February/000249.html Mailing list] discussion | |||
== 15th Feb 2012 == | |||
Tried two new DNA extraction protocols (Chelex and Meat tenderiser). The problem with both of these protocols is that we have no idea how much DNA we are actually extracting. Perhaps we are extracting too much. "There is really no lower limit," says Bugs, because PCR will amplify the DNA. | |||
More details: [[/results-20120215]] | |||
== 22nd Feb 2012 == | |||
Tonight we did DNA extraction following the protocols from the 15th -- 4 people did a meat tenderiser and alcohol-based extraction, and 3 did a heating and Chelex-based extraction. | |||
We followed the recommendations from bugs for primer strength and overall reaction volume (see the notes from the 15th). | |||
We set the thermocycler to the program mentioned in a paper which used the same primers (see notes from the 12th): | |||
Denature: 1 minute at 94 C; Anneal: 2 minutes at 65 C; Extend: 3 minutes at 72 C. | |||
Unfortunately we ran out of time and did not wait for the thermocycler to complete for visualisation. we will do that next week. | |||
== 29th Feb 2012 == | |||
This week we ran a gel for the results of last week's work. Buffer was ~20g of 10xTBE diluted with ~200g of battery top-up water. Gel was made with ~1.0g of agarose in ~100g of buffer, and 5u of EthBr added. We judged how much to pour into the tray by eye, but when the wells came up very shallow, we thought maybe use the scales in future. | |||
For whatever reason, the experiment was not a success. We couldn't visualise anything. The ladder was a barely-discernible smear, and none of the other lanes was visible at all. | |||
We decided to do another run: Nicholas, Helena, Mike, and Tonderai contributed DNA for overnight PCR. | |||
== | == 1st March 2012 == | ||
Started using orange filter for gel visualizations. | |||
'''Visualised the previous night's PCR run:''' | |||
[[File:2012-02-29-results-1.JPG|200px|thumb|none|Results of experiment 1. From top: Empty lane, ladder, Nicholas, Helena, Mike, Tonderai]] | |||
[[ | |||
It is hard to see, but it almost looks like lane 4 (Tonderai? Or was it Mike?) is almost right -- there seem to be two distinct bands. | |||
We did not cover the master mix / primers / template solution with oil. The PCR reaction vessels had condensed liquid all over the vessel, including up near the lid. I (Nicholas) theorise that the mixture evaporated, inhibiting PCR, in some cases severely. | |||
'''Attempt to remedy evaporation during PCR:''' | |||
I tried two different oils: | |||
* "Laser Lube" (Thanks to Hipster for this description) -- machine lubricant, from a squeeze bottle near a drill press | |||
* A mysterious bottle of oil I found in the biohacking box, which could be mineral oil (though it's not the same bottle that we were using before). | |||
[[ | [[File:2012-03-01-results-1.JPG|200px|thumb|none|Results of experiment 1 From top: Empty lane, ladder, Nicholas - meat tenderiser and laser lube, Nicholas - meat tenderiser and mystery oil, Nicholas - heat to lyse and mystery oil]] | ||
Once again, two distinct bands are visible, but they don't seem to be in the right places on the gel. | |||
I conclude that we can use either mystery oil or laser lube. | |||
We continued to run the gels with visualisation once every ~12 mins | |||
[[File:Gel_IMG_20120301_194156_edit.jpg|200px|thumb|none| At about 19:42, we have all six lines of the ladder and something that looks like a line in the third lane somewhere about the 3rd or 4th rung on the ladder (which is what we are hoping for)]] | |||
[[/results- | More details: [[/results-20120301]] | ||
== | == 7th March 2012 == | ||
Data lost | |||
== | == 11th March 2012 == | ||
'''Extraction | Two separate DNA extraction protocols: Chelex and meat tenderiser. | ||
Chelex: rinse mouth with salt. Spit small amount of liquid into tube. Add 500ul Chelex. Incubate at 98 degrees for 6 minutes. Centrifuge for 5 minutes. Take 10ul supernatant. | |||
Meat tenderiser: rinse mouth with salt. Spit small amount of liquid into tube. Add equal amount of detergent. Vortex for 30 seconds. Add a few grains meat tenderiser. Vortex for 30 seconds. Allow to sit for several seconds. Add a small amount of rubbing alcohol. Observe DNA strands precipitating out of solution. Take 10ul of liquid and DNA strands. Incubate at 70 degrees for several minutes. Add 5 ul water. | |||
PCR was set at D=94 degrees, 1 minute; A=57 degrees, 1 minute; E = 72 degrees, 1 minute. I used 5 ul of sample, 6 ul of primers, 12.5 ul of Taq master mix. | |||
Result was visualised on 1% agarose gel stained with 2ul/40ml of ethidium bromide under 300nm UV. DNA ladder was visible. Both samples showed a faint blur running ahead of the ladder and no other information. | |||
I conclude that the DNA extraction technique is inadequate. | |||
Detailed thoughts on what might be going wrong: [[/results-20120311]] | |||
== 21st March 2012 : Success!! == | |||
Following [http://www.jicef.or.jp/wahec/ful318.htm this chelex protocol] | |||
* Chelex 5ul | |||
* PCR at 96x1 58x1 72x1 @ 35 cycles - with initial denat at 94 for 2 mins | |||
* Oil on chelex durin incubation | |||
* 8ul of 2.5uM primer (4ul of each, mixed) | |||
[[File:GEL_fuck_yes.jpg|400px|thumb|none|The moment of truth]] | |||
[https://lists.kentgeek.org/pipermail/lhs-biohacking/2012-March/000299.html The announcement on the mailing list] | |||
== 22nd March 2012: Anti-success == | |||
After such a great result yesterday it was disappointing to see a non-result today: | |||
[[File:Biohacking_20120322_result.jpg|400px|thumb|none|No result. Extreme left is (warped) ladder, other wells show only primer bands extending way beyond the ladder]] | |||
It's interesting in itself because we used a positive control thanks to Tom, plus known-good primers (also thanks to Tom). This suggests that the problem with intermittent results is the physical PCR process itself, rather than DNA extraction. (The previous good result already suggested that the primers and master mix weren't to blame.) | |||
== 27th March 2012 == | |||
Meat tenderiser extraction: 35 cycles of D - 96° for 1:30, A - 55° for 2:00, E - 72° for 3:00. No result. | |||
More detailed info in the lab book. | |||
== 28th March 2012 == | |||
Expt. 1: Chelex extraction: 35 cycles of D - 96° for 1:00, A - 58° for 2:00, E - 72° for 3:00. No result. | |||
Expt. 2: Chelex extraction: 35 cycles of D - 96° for 5:00, A - 45° for 5:00, E - 72° for 5:00. No result. | |||
More detailed info in the lab book. | |||
== 4th April 2012 == | |||
Mixed extractions: 35 cycles of D - 94° for 1:00, A - 50° for 0:30, E - 72° for 1:00. Mostly good results. Detailed info [https://lists.kentgeek.org/pipermail/lhs-biohacking/2012-April/000318.html here]. | |||
== 8th April 2012 == | |||
Meat tenderiser extraction: 35 cycles of D - 94° for 1:00, A - 55° for 1:00, E - 72° for 1:00. No result. | |||
More detailed info in the lab book. | |||
== 11th April 2012 == | |||
Mixed extractions: 35 cycles of D - 94° for 1:00, A - 54° for 0:30, E - 72° for 1:00. No result. | |||
More detailed info in the lab book. | |||
== 18th April 2012 == | |||
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. Good result. Only Nicolas' DNA used. | |||
More detailed info in the lab book. | |||
== 25th April 2012 == | |||
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. Good result. Detailed info [https://lists.kentgeek.org/pipermail/lhs-biohacking/2012-April/000352.html here]. | |||
The major problem seems to be quality of the extracted DNA: PCR product is frequently quite faint. As of April 26, 2012, DNA extraction technique will be the main focus for the amelogenin test (and for genetic testing of human DNA in general). | |||
== 16th May 2012 == | |||
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. No result. | |||
More detailed info in the lab book. | |||
== 19th June 2012 == | |||
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 54° for 0:30, E - 72° for 1:00. No result. | |||
More detailed info in the lab book. | |||
== 1st August 2012 == | |||
Made a gel. 0.5g of agar. ~5g TBE. ~45g water (which sort?) 2.5ul. | |||
More detailed info in the lab book. | |||
= Complete protocol as of June 2012 = | |||
==Extraction== | |||
* Fill a PCR tube with 250 ul of 5% Chelex solution. | * Fill a PCR tube with 250 ul of 5% Chelex solution. | ||
| Line 233: | Line 378: | ||
* Centrifuge and take supernatant. | * Centrifuge and take supernatant. | ||
==PCR== | |||
* Use the primers 5'-CTGATGGTTGGCCTCAAGCCTGTG-3' and 5'-TAAAGAGATTCATTAACTTGACTG-3' | * Use the primers 5'-CTGATGGTTGGCCTCAAGCCTGTG-3' and 5'-TAAAGAGATTCATTAACTTGACTG-3' | ||
| Line 240: | Line 385: | ||
* Then 30 cycles of D = 96 C for one minute, A = 54 C for 30 seconds, E = 72 C for one minute. | * Then 30 cycles of D = 96 C for one minute, A = 54 C for 30 seconds, E = 72 C for one minute. | ||
==Electrophoresis== | |||
* Prepare a 1% agarose gel using 1X TBE and 2ul / 40ml of ethidium bromide. | * Prepare a 1% agarose gel using 1X TBE and 2ul / 40ml of ethidium bromide. | ||
* Add 1 part loading buffer to 5 parts sample | |||
* Electrophorese at between 80v and 100v for 30 to 60 minutes. | * Electrophorese at between 80v and 100v for 30 to 60 minutes. | ||
==Visualisation== | |||
* Wear glasses and long sleeves. | * Wear glasses and long sleeves. | ||
* Visualise using a 300nm UV light held above the gel. View (or photograph) through an orange filter. | * Visualise using a 300nm UV light held above the gel. View (or photograph) through an orange filter. | ||
= Future Work = | |||
Given that we get this working, it should provide a basis for further experiments. As far as I understand it, it is possible to apply this sort of technique, with appropriate primers, to do detection of SNPs [http://en.wikipedia.org/wiki/Single-nucleotide_polymorphism] opening up the possibility of testing for something common like [http://en.wikipedia.org/wiki/RHD_%28gene%29 RHD] or [http://en.wikipedia.org/wiki/Melanocortin_1_receptor MC1R] alleles, and perhaps providing a resource for people who want to genetically test themselves for rarer things a la Katherine Aull (but it is much simpler and cheaper to get such tests run commercially, and there is a difficult ethical case to answer about providing genetic testing without proper expert medical support and interpretation of results). | Given that we get this working, it should provide a basis for further experiments. As far as I understand it, it is possible to apply this sort of technique, with appropriate primers, to do detection of SNPs [http://en.wikipedia.org/wiki/Single-nucleotide_polymorphism] opening up the possibility of testing for something common like [http://en.wikipedia.org/wiki/RHD_%28gene%29 RHD] or [http://en.wikipedia.org/wiki/Melanocortin_1_receptor MC1R] alleles, and perhaps providing a resource for people who want to genetically test themselves for rarer things a la Katherine Aull (but it is much simpler and cheaper to get such tests run commercially, and there is a difficult ethical case to answer about providing genetic testing without proper expert medical support and interpretation of results). | ||
We can also test for nonhuman DNA, i.e. non-genetic diseases caused by viruses or bacteria; test the provenance of various foods (fish in general and sashimi in particular would seem like a good option); or attempt DNA fingerprinting to identify particular people (I haven't looked into this but would guess it involves a few different primers targeting highly-variable portions of the genome). | We can also test for nonhuman DNA, i.e. non-genetic diseases caused by viruses or bacteria; test the provenance of various foods (fish in general and sashimi in particular would seem like a good option); or attempt DNA fingerprinting to identify particular people (I haven't looked into this but would guess it involves a few different primers targeting highly-variable portions of the genome). | ||
[[Category:Biohacking]] | |||
[[Category:Projects]] | |||
Latest revision as of 00:17, 25 April 2014
General
The general plan, with thanks to Maria, is: obtain dna samples, extract dna, pcr dna with amelogenin primers, and then run that on agarose gel.
There is still work that could be done, checking the legals, ethics and safety. eg: [1] [2] [3]
I figure we try to arrive at a definitive list of stuff (bill of materials) we need for the experiment, and source the bits we still need, perhaps avoiding unnecessary multiple delivery charges.
There is also a need to figure out how long this will all take and schedule it. The overall duration from start to finish is going to be quite long.
It might help to try out some parts, such as the extraction, before hand, although I don't see how we know whether that works until we run it through the rest. We could practise making gels, running a ladder on them, to figure out mixtures and timings and practise with detecting the dye.
Sampling
Current plan is to sterilise plastic sticks for consenting adult volunteers to rub inside their cheeks. Legally (I am so not a lawyer, and this is not legal advice), reading them a written spiel and getting their verbal agreement with witnesses and seeing them take the sample seems to be sufficient , although the suggestion has been made that a simple signed form might help avoid a possible problem with forgetful witnesses.
cotton swabs are another possible option.
Extraction
There are a number of written protocols out there
and a few videos on youtube
This seems to be the typical hackers version [6] which is basically soap to lyse the cells and then meat tenderiser as a protease and separation using alcohol. Reagents are all commonly available, so seems safe enough. will this will work with PCR?
Katherine Aull was boiling hers to get lysis and denature the proteins.
do we also need to do DNA precipitation? [7] why?
The sampling and extraction are crucial steps if the following steps are to work. dna contamination can be amplified in the pcr stage. there is a suggestion that there is an amount of black art in getting this to work well. It might repay the effort to try several different extractions and run them side by side for comparison.
PCR
[8] File:Polymerase.pdf Media:DNA_Ladders.jpg Media:New_ladder.jpg
paper more-or-less at random, Eng 1994 where it gives:
PCR Amplification of the Amelogenin Locus The primers used to amplify the X and Y amelogenin sequences were previously described by Nakahori and colleagues [6]. The primer sequences are as follows: primer AMXY-1F (5'-CTGATGGTTGGCCTCAAGCCTGTG-3') primer AMXY-2R (5'-TAAAGAGATTCATTAACTTGACTG-3') PCR was conducted using the Perkin-Elmer GeneAmp | PCR System 9600 Thermal Cycler. Genomic DNA samples were amplified in a 100 txL reaction volume containing 0.2 mM of each dNTP, 50 mM KCI, 10 mM Tris.HC1 pH 8.3, 4.0 mM MgC12, 120 pmoles of each primer, and 2.5 U Taq polymerase. PCR was run for 30 cycles of 94~ for 1 rain (denature), 65~ for 2 min (anneal), and 72~ for 3 min (extend). Following amplification, the PCR products were analyzed by electrophoresis on 1.2% agarose gels run with 1 X TBE (89 mM Tris borate, 89 mM boric acid, 2 mM EDTA) or 6% nondenaturing polyacryl- amide gels run with 1 X TBE. The gels were stained with ethidium bromide and the fragments were visualized by fluorescence under ultraviolet light.
Andy suggests that we go with the instructions on our polymerase for the PCR.
S4438 (with SYBR green)[9]
D6442 (without)[10]
There is a question mark over whether we need a smaller pippette, or perhaps we can let out some solutions to use a larger volume. Katherine Aull was using syringes.
We need tubes for the thermal cycler.
We have SYBR green to replace the ethidium bromide.
Gel electrophoresis
Procedure
Eng has
1.2% agarose gels run with 1 X TBE (89 mM Tris borate, 89 mM boric acid, 2 mM EDTA)
If our dye is SYBR Green I then we need blue light (λmax = 497 nm)
It seems we need to manufacture new electrophoresis equipment or has someone taken it to be repaired? I looked into the possibility of making the gel tray as a single bent piece, but the guys I know said they couldn't do it on 5mm acetate.
We are still looking for a better alternative to the copper electrode that gets eaten by the reagents.
It is common to run reference samples (ladders) alongside the samples, so as to see that weight of the sample. would also be handy to calibrate this process first. ladders can be bought, do we already have?
So, we need TAE or TBE for the buffer.
Tris Safety Data Sheet Boric Acid Safety sheet Example COSHH for PCR and gel electrophoresis with TAE
Stains
SafeView and SafeWhite are from NBS Bio. There are manuals and safety sheets there.
interesting doc about ethidium bromide disposal in th uk here: [11]
What we are looking for
We expect bands at X = 977 bp and Y = 788 bp

on paper
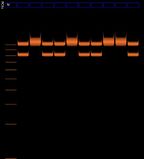
example from cybertory
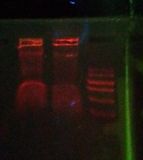
Is this it? see /results-20120301

"FUCK YES" see /results-20120321




It seems that the lines at X=977bp are in different positions in the male and female lanes in the cybertory example. discuss.
Results
Individuals with deletions such that this test doesn't work on them are extremely rare. So we hope to get correct results for all samples. We discussed blinding the samples to avoid possible bias in interpretation reading the gel, but if we can photograph the gel that could serve that purpose.
It might be nice to do video recording and a write up.
First gel run, Dec 7, 2011
On this date, we had not yet received our PCR tubes, so we were unable to do PCR. Instead, we decided to test our ability to do electrophoresis, using a DNA ladder and SafeWhite stain. We followed the following protocol:
* Agarose, added to buffer, microwaved. 1 gram of agarose to 100ml of tbe: 1% agarose mix will work for 0.4 to 7kb * cool in a water bath set at 50-55c. * add 5 ul of ethidium bromide to gel if using ethidium bromide. If using safewhite add nothing yet * Add gel to mold: cover sides with masking tape * Add comb to gel * Put in fridge for half an hour (or, basically: cool until completely set) * remove comb. * Place gel, with tray, in electrophoresis box. * Fill electrophoresis box with TBE, to cover the gel. * prepare sample: 5ul of ladder and 2 ul of safewhite (for the 6x solution) * add sample to gel tray. * electrophorese at 100v for 1 hour. Negative terminal closest to DNA * Visualise under UV at 280-300nm.
The reference I used for this protocol: http://www.biocompare.com/protocols/protocol/318/Agarose-Gel-Electrophoresis.html
Unfortunately we were unable to detect any green glow which would indicate that the DNA in the ladder had bound to the Safewhite.
We resolved to make the next test a simple visualisation test: combine ladder with Safewhite and attempt to visualise under UV.
SafeWhite test, Dec 9, 2011
Nicholas combined 2.5ul SafeWhite and 2.5ul DNA Marker-D. He was unable to make this mixture glow under UV.
Second gel run, Dec 13, 2011
Nicholas performed the same protocol as in the "First gel run, Dec 7, 2011", with the following changes:
* SafeWhite was not added. * Two wells were loaded with 3ul of DNA Marker-D. * After electrophoresis, the gel was cut in half, with one active well per half. * One half was submerged in 0.002% methylene blue solution (in 0.1X TBE) for one hour. * The other half was submerged in 0.001% gentian violet solution (in 0.1X TBE) for half an hour. * No staining was observed.
3rd or 4th gel run, Jan 25, 2012
We successfully visualised a ladder using the following protocol:
Quantities of chemicals:
- 1% TBE, 1 part "TBE 10x" to 9 parts water
- Agarose, 1 gram per 100ml of TBE
- Ethidium bromide stock solution, 1 gram of powdered EtBr per 100ml of TBE
- 2ul of ethidium bromide stock solution per 40ml of agarose
Protocol:
- 200ml of agarose: 2 grams of agarose powder to 200ml of 1% TBE solution
- Microwaved the agarose, cooled to 50 degrees, added 10ul of ethidium bromide stock solution
- Poured the agarose into a mould, trying to keep the gel as thin as possible. We ended up using about 100ml of agarose. Waited for agarose to cool.
- Added 6ul of a newly-ordered DNA ladder into two wells.
- Electrophoresed, 100 volts for half an hour (thereabouts -- power supply was current limited to 0.8 amps, so voltage dropped to about 80 volts as the electrophoresis progressed)
- Visualised by placing a fluorescent tube with peak emission at 300 nm directly underneath the gel.
1st Feb 2012
our Technical datasheet gives numbers for water to add to rehydrate our primers to 100uM.
8th Feb 2012

Nice picture

not such a good image

after crop and contrast stretch

Everybody's looking for the ladder

after contrast stretch
after desaturate and contrast stretch

hmm...







12th Feb 2012

Cell lysis.

Equipment for PCR and electrophoresis.

After 20 minutes of electrophoresis bands were observed in lanes 2, 3, 4, 6, and 7. The ladder was also visible in lane 5 but can't be seen in this photo.

After 45 minutes of electrophoresis the bands had become very blurry.
After 45 minutes of electrophoresis, close-up. The ladder is faintly visible.
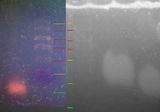
On the left is the gel from 8th Feb 2012. The lines in red are clearly lines in the ladder, the orange lines are putative ladder (less certain), and the green lines are the bands. If the interpretation presented here is correct, then this result repeats the results of last Wednesday.






Discussion:
- The bands are very blurry and wonky, as if the agar was of varying densities throughout.
- Bands on lanes 3, 4, and 6 travelled less far than bands on lanes 2 and 7. Lane 2 did not include template (i.e. DNA); and I ran out of template in lane 7. So this is an encouraging result.
- The bands are within the bounds of the ladder this time.
- Bands on lanes 3, 4, and 6 are more blurry than bands on lanes 2 and 7. One exciting theory is that bands on those lanes actually worked, and the blurriness is because of the 200 base-pair difference between the two amelogenin sequences; whereas the bands on lanes 2 and 7 contain un-used primer and master mix, which is a) shorter sequences and b) more well-defined (presumably because there is less of a difference between the lengths).
More details on the days results and our discussions: /results-20120212
Mailing list discussion
15th Feb 2012
Tried two new DNA extraction protocols (Chelex and Meat tenderiser). The problem with both of these protocols is that we have no idea how much DNA we are actually extracting. Perhaps we are extracting too much. "There is really no lower limit," says Bugs, because PCR will amplify the DNA.
More details: /results-20120215
22nd Feb 2012
Tonight we did DNA extraction following the protocols from the 15th -- 4 people did a meat tenderiser and alcohol-based extraction, and 3 did a heating and Chelex-based extraction.
We followed the recommendations from bugs for primer strength and overall reaction volume (see the notes from the 15th).
We set the thermocycler to the program mentioned in a paper which used the same primers (see notes from the 12th):
Denature: 1 minute at 94 C; Anneal: 2 minutes at 65 C; Extend: 3 minutes at 72 C.
Unfortunately we ran out of time and did not wait for the thermocycler to complete for visualisation. we will do that next week.
29th Feb 2012
This week we ran a gel for the results of last week's work. Buffer was ~20g of 10xTBE diluted with ~200g of battery top-up water. Gel was made with ~1.0g of agarose in ~100g of buffer, and 5u of EthBr added. We judged how much to pour into the tray by eye, but when the wells came up very shallow, we thought maybe use the scales in future.
For whatever reason, the experiment was not a success. We couldn't visualise anything. The ladder was a barely-discernible smear, and none of the other lanes was visible at all.
We decided to do another run: Nicholas, Helena, Mike, and Tonderai contributed DNA for overnight PCR.
1st March 2012
Started using orange filter for gel visualizations.
Visualised the previous night's PCR run:

It is hard to see, but it almost looks like lane 4 (Tonderai? Or was it Mike?) is almost right -- there seem to be two distinct bands.
We did not cover the master mix / primers / template solution with oil. The PCR reaction vessels had condensed liquid all over the vessel, including up near the lid. I (Nicholas) theorise that the mixture evaporated, inhibiting PCR, in some cases severely.
Attempt to remedy evaporation during PCR:
I tried two different oils:
- "Laser Lube" (Thanks to Hipster for this description) -- machine lubricant, from a squeeze bottle near a drill press
- A mysterious bottle of oil I found in the biohacking box, which could be mineral oil (though it's not the same bottle that we were using before).

Once again, two distinct bands are visible, but they don't seem to be in the right places on the gel.
I conclude that we can use either mystery oil or laser lube.
We continued to run the gels with visualisation once every ~12 mins

More details: /results-20120301
7th March 2012
Data lost
11th March 2012
Two separate DNA extraction protocols: Chelex and meat tenderiser.
Chelex: rinse mouth with salt. Spit small amount of liquid into tube. Add 500ul Chelex. Incubate at 98 degrees for 6 minutes. Centrifuge for 5 minutes. Take 10ul supernatant.
Meat tenderiser: rinse mouth with salt. Spit small amount of liquid into tube. Add equal amount of detergent. Vortex for 30 seconds. Add a few grains meat tenderiser. Vortex for 30 seconds. Allow to sit for several seconds. Add a small amount of rubbing alcohol. Observe DNA strands precipitating out of solution. Take 10ul of liquid and DNA strands. Incubate at 70 degrees for several minutes. Add 5 ul water.
PCR was set at D=94 degrees, 1 minute; A=57 degrees, 1 minute; E = 72 degrees, 1 minute. I used 5 ul of sample, 6 ul of primers, 12.5 ul of Taq master mix.
Result was visualised on 1% agarose gel stained with 2ul/40ml of ethidium bromide under 300nm UV. DNA ladder was visible. Both samples showed a faint blur running ahead of the ladder and no other information.
I conclude that the DNA extraction technique is inadequate.
Detailed thoughts on what might be going wrong: /results-20120311
21st March 2012 : Success!!
Following this chelex protocol
- Chelex 5ul
- PCR at 96x1 58x1 72x1 @ 35 cycles - with initial denat at 94 for 2 mins
- Oil on chelex durin incubation
- 8ul of 2.5uM primer (4ul of each, mixed)
The announcement on the mailing list
22nd March 2012: Anti-success
After such a great result yesterday it was disappointing to see a non-result today:

{kind=link}
{kind=link}
It's interesting in itself because we used a positive control thanks to Tom, plus known-good primers (also thanks to Tom). This suggests that the problem with intermittent results is the physical PCR process itself, rather than DNA extraction. (The previous good result already suggested that the primers and master mix weren't to blame.)
27th March 2012
Meat tenderiser extraction: 35 cycles of D - 96° for 1:30, A - 55° for 2:00, E - 72° for 3:00. No result.
More detailed info in the lab book.
28th March 2012
Expt. 1: Chelex extraction: 35 cycles of D - 96° for 1:00, A - 58° for 2:00, E - 72° for 3:00. No result.
Expt. 2: Chelex extraction: 35 cycles of D - 96° for 5:00, A - 45° for 5:00, E - 72° for 5:00. No result.
More detailed info in the lab book.
4th April 2012
Mixed extractions: 35 cycles of D - 94° for 1:00, A - 50° for 0:30, E - 72° for 1:00. Mostly good results. Detailed info here.
8th April 2012
Meat tenderiser extraction: 35 cycles of D - 94° for 1:00, A - 55° for 1:00, E - 72° for 1:00. No result.
More detailed info in the lab book.
11th April 2012
Mixed extractions: 35 cycles of D - 94° for 1:00, A - 54° for 0:30, E - 72° for 1:00. No result.
More detailed info in the lab book.
18th April 2012
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. Good result. Only Nicolas' DNA used.
More detailed info in the lab book.
25th April 2012
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. Good result. Detailed info here.
The major problem seems to be quality of the extracted DNA: PCR product is frequently quite faint. As of April 26, 2012, DNA extraction technique will be the main focus for the amelogenin test (and for genetic testing of human DNA in general).
16th May 2012
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 56° for 0:30, E - 72° for 1:00. No result.
More detailed info in the lab book.
19th June 2012
Chelex extraction: 35 cycles of D - 96° for 1:00, A - 54° for 0:30, E - 72° for 1:00. No result.
More detailed info in the lab book.
1st August 2012
Made a gel. 0.5g of agar. ~5g TBE. ~45g water (which sort?) 2.5ul.
More detailed info in the lab book.
Complete protocol as of June 2012
Extraction
- Fill a PCR tube with 250 ul of 5% Chelex solution.
- Scrape cheek cells using a pipette tip.
- Stir the pipette tip in the Chelex so that the cells are transferred. Perhaps also spit down the pipette tip.
- Incubate at 56 C for 30 minutes. Vortex for a few seconds. Incubate at 94 C for 10 minutes.
- Centrifuge and take supernatant.
PCR
- Use the primers 5'-CTGATGGTTGGCCTCAAGCCTGTG-3' and 5'-TAAAGAGATTCATTAACTTGACTG-3'
- Use 12.5 ul of Taq master mix, 2 ul of each primer, 3 ul of template, and 5.5 ul of distilled water.
- Initial denat. of 96 C for 5 minutes.
- Then 30 cycles of D = 96 C for one minute, A = 54 C for 30 seconds, E = 72 C for one minute.
Electrophoresis
- Prepare a 1% agarose gel using 1X TBE and 2ul / 40ml of ethidium bromide.
- Add 1 part loading buffer to 5 parts sample
- Electrophorese at between 80v and 100v for 30 to 60 minutes.
Visualisation
- Wear glasses and long sleeves.
- Visualise using a 300nm UV light held above the gel. View (or photograph) through an orange filter.
Future Work
Given that we get this working, it should provide a basis for further experiments. As far as I understand it, it is possible to apply this sort of technique, with appropriate primers, to do detection of SNPs [12] opening up the possibility of testing for something common like RHD or MC1R alleles, and perhaps providing a resource for people who want to genetically test themselves for rarer things a la Katherine Aull (but it is much simpler and cheaper to get such tests run commercially, and there is a difficult ethical case to answer about providing genetic testing without proper expert medical support and interpretation of results).
We can also test for nonhuman DNA, i.e. non-genetic diseases caused by viruses or bacteria; test the provenance of various foods (fish in general and sashimi in particular would seem like a good option); or attempt DNA fingerprinting to identify particular people (I haven't looked into this but would guess it involves a few different primers targeting highly-variable portions of the genome).